Our technology and evolutionary molecular engineering
Biopolymers such as nucleic acids (DNA/RNA) and proteins have been gaining the high functions through 4
billion years of history. Nowadays, researchers are able to synthesize the DNA, RNA, and protein with their
purpose sequences. However, designing nucleic acids and proteins for the purpose function, in other words,
structurally approaching molecular design is very hard yet. Evolutionary molecular engineering is the
technology which evolves the molecules under the lab setting with high speed, and enables the evolved
molecules which are new functional biopolymers.
In EME, we use one of the evolutionary molecular engineering techniques; cDNA display method as our core
technology. cDNA display method allows us to gain specific and high affinity (up to nM level of KD) VHH
antibody and cyclic peptide by high-throughput screening platform (it has been developed by EME), and these
candidates are applied for innovation of new medications.
The history of developing cDNA display method
Epsilon Molecular Engineering Inc. was founded as a biotechnology start-up company emerged from Saitama
university which has used the core technology of evolutionary molecular engineering. From 1985, Saitama
university, Prof. Hushimi and his team have been researching evolutionary molecular engineering.
Recently, evolutionary molecular engineering has been highlighted in the world. For instance, Prof. George
P. Smith (He received the Norvel Prize of Chemistry in 2018) has started researching the Phage display
method for peptides and antibodies. In vitro virus (mRNA display) method which is one of the cell free genotype/phenotype linking systems
developed first in the world by Dr. Nemoto and Prof. Hushimi in 1997. mRNA display method has ten thousand
times efficiency than phage display method. However, the mRNA display method is less stable than other
methods. Thus, we developed cDNA display method as genotype-phenotype linking strategy which has much larger
diversity and higher quality. The cDNA display method can be replaceable for mRNA display because of its
stability.
VHH / Peptide
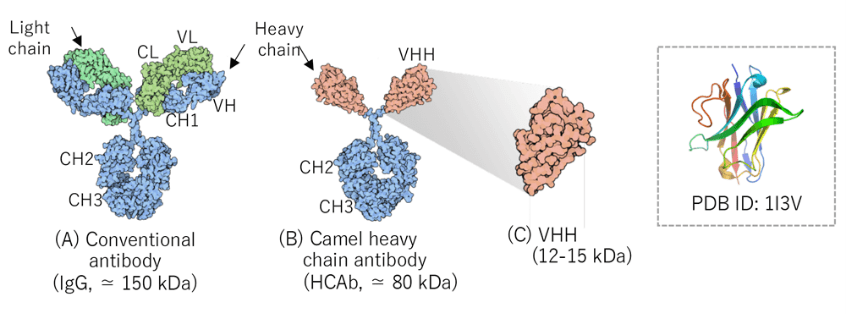
What is VHH?
Camelids such as alpacas (Vicugna pacos) and llamas (Lama glama) produce not only IgG antibodies consisting
of heavy chains (H chains) and light chains (L chains) (A) but also antibodies consisting of only heavy
chains (heavy-chain antibodies, HCAb) (B). The single variable domain of HCAb is called VHH with a molecular
weight of 12−15 kDa (C). VHH has high antigen specificity and affinity like that of a conventional IgG
antibody. While an IgG antibody forms antigen-binding sites with the three complementarity determining
regions (CDRs) of each of VH and VL, VHH recognizes antigens with three CDRs. CDR3, in particular, is an important region for antigen binding. VHH preferentially
recognizes clefts and cavities in the target molecules.
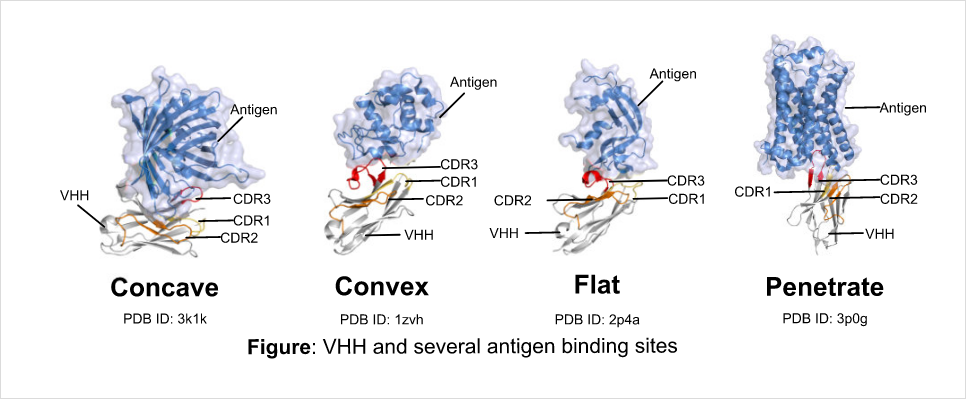
VHH binds to several types of epitopes
Structure of antigen binding site (paratope) which CDR of VHH form have much larger diversity than
conventional antibodies. Conventional antibodies bind to the convex areas of antigen. On the other hand, VHH
favors to bind to hollows and gaps of antigen, but also binds to the convex areas and nearly flat surface
structures of antigen. Moreover, VHH recognizes the low molecular particles.
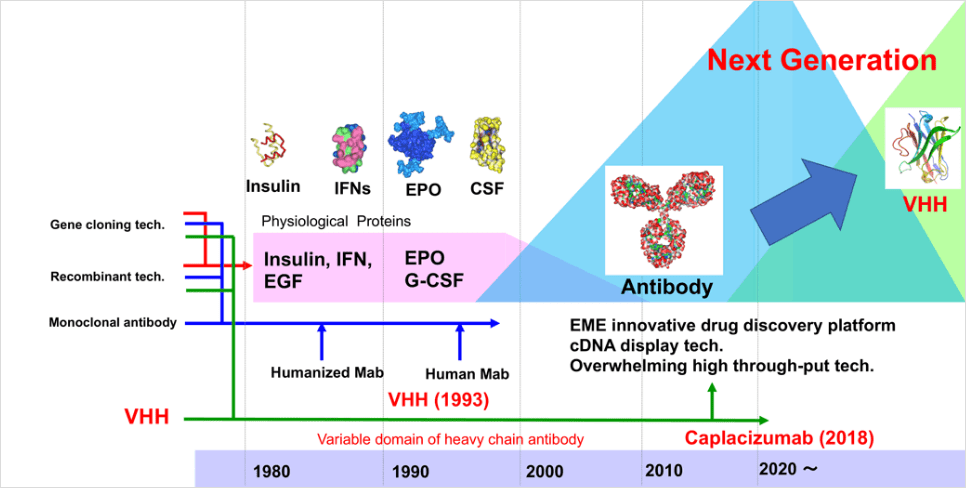
Paradigm shift in biopharmaceutical developments: appearance of VHH
Since the 1980s, biotechnology-based recombinant proteins, such as insulin and erythropoietin, have been
developed as new medications. Subsequently, in 1987, the first humanized antibody technology was developed
then the antibody-drug discovery got the spotlight as next-generation biomedicines. Research and development
of antibodies against many target molecules have been promoted along with the development of genome-based
drug discovery since the 1990s, and antibody drugs accounted for 62% of the biopharmaceutical market in
2018. While antibody drugs are expected to be developed as the next-generation antibodies in the future
based on innovative technology and concepts, we have faced the limitations of antibody drug industry.
Conversely, VHH was discovered in 1993 by Prof. Hamers at Vrije Universiteit Brussel in
Belgium(Hamers-Casterman, C., et al. 1993). Although VHH has many features favoring drug discovery, major
pharmaceutical companies did not consider VHH as an attractive new biomolecule in the 1990s because VHH was
discovered at the same time when antibody drugs attracted much attention as next-generation
biopharmaceuticals. However, a biotech start-up company started aiming to commercialize VHH in Belgium as
one of the low-molecular antibody drugs, then research and development of VHH were started. In 2018, FDA
approved Caplacizumab as the first VHH antibody drug against von Willebrand Factor for the treatment of
acquired thrombotic thrombocytopenic purpura. After that, many of VHH antibody medications continuously have
been moving to clinical trial phases, and VHH has been highlighted as one of the next generation
biomolecules for new drug discovery.
Cyclic peptides/peptide aptamers
In EME, we have a library of cyclic peptides. The greatest feature of this cyclic peptide library is that we
synthesize the cyclic peptides with disulfide linkage or chemical linkage agents between pairs of cysteines.
These amino acids of cyclic peptides are randomized. Thus, we can have several lengths and sequences of
cyclic peptides. For more information, please see our page “Library”.
Also, we synthesize cyclic peptides with linkage between two cysteines using thiol group (disulfide linkage)
or a chemical linkage agent. Based on these methods, we have been improving efficiency of forming a cDNA
display library for screening cyclic peptides.